
Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
Lissencefalija možganov
Medicinski strokovnjak članka

Zadnji pregled: 12.07.2025
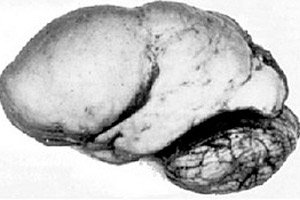
Med organskimi možganskimi patologijami izstopa prirojena anomalija razvoja možganov, kot je lisencefalija, katere bistvo je v skoraj gladki površini skorje njenih hemisfer - z nezadostnim številom zvitkov in brazd. [ 1 ]
V popolni odsotnosti zvitkov se opredeli agirija, prisotnost več širokih ravnih zvitkov pa se imenuje pahigirija. Te okvare, tako kot nekatere druge redukcijske deformacije možganov, imajo v ICD-10 kodo Q04.3.
Epidemiologija
Glede na statistiko redkih bolezni je 1–1,2 primera lisencefalije na 100 tisoč novorojenčkov. [ 2 ], [ 3 ]
Po nekaterih podatkih je pri otrocih z Miller-Diekerjevim sindromom do 25–30 % primerov klasične lisencefalije; točkovne mutacije in delecije genov LIS1 in DCX so odkrite pri skoraj 85 % bolnikov. [ 4 ]
Genetske študije 17 genov, povezanih z lisencefalijo, so pokazale, da mutacija ali delecija LIS1 predstavlja 40 % bolnikov, 23 % pa jih je povezanih z mutacijo DCX, sledita pa TUBA1A (5 %) in DYNC1H1 (3 %).[ 5 ]
Vzroki lissencefalija
Vsi znani vzroki za nastanek možganske skorje (cortex cerebri) skoraj ali popolnoma brez zvitkov in brazd, ki povečajo "delovno območje" človeških možganov in zagotavljajo "produktivnost" centralnega živčnega sistema, so povezani z motnjami v njegovem perinatalnem razvoju. To pomeni, da se pri plodu razvije lisencefalija. [ 6 ]
Neuspeh pri tvorbi plasti možganske skorje ploda pri lisencefaliji je posledica nenormalne migracije nevronov, ki jo tvorijo, ali prezgodnje prekinitve tega procesa.
Ta proces, ki je bistven za cerebrokortikalno histogenezo, poteka v več fazah od 7. do 18. tedna nosečnosti. Glede na povečano občutljivost na genetske mutacije, pa tudi na različne negativne fizikalne, kemične in biološke vplive, lahko vsako odstopanje od norme povzroči napačno lokalizacijo nevronov z morebitnim nastankom odebeljene plasti sive snovi skorje brez značilne strukture. [ 7 ]
V nekaterih primerih je lisencefalija pri otrocih povezana s sindromi Miller-Dieker, Walker-Warburg ali Norman-Roberts.
Preberite tudi – Razvojne napake možganov
Dejavniki tveganja
Poleg mutacij v nekaterih genih so dejavniki tveganja za rojstvo otroka s tako hudo okvaro še kisikovo stradanje (hipoksija) ploda; nezadostna prekrvavitev možganov (hipoperfuzija); akutna možganska kap v obliki perinatalne kapi; patologije posteljice; virusne okužbe nosečnice (vključno s TORCH); [ 8 ] težave s splošnim metabolizmom in delovanjem ščitnice; kajenje, alkohol, psihotropne in narkotične snovi; uporaba številnih zdravil; povečana raven sevanja. [ 9 ]
Patogeneza
Vsi primeri lisencefalije nimajo patogeneze, ki bi jo povzročile kromosomske nepravilnosti in genske mutacije. Vendar pa so znani nekateri geni, ki kodirajo beljakovine, ki igrajo pomembno vlogo pri pravilnem gibanju nevroblastov in nevronov vzdolž radialnih glialnih celic – da tvorijo možgansko skorjo. In mutacije teh genov vodijo do te patologije. [ 10 ]
Predvsem gre za sporadične mutacije (brez dednosti) gena LIS1 na kromosomu 17, ki uravnava citoplazemski motorični protein mikrotubulov dinein, pa tudi gen DCX na kromosomu X, ki kodira protein dvojni kortin (lisencefalin-X). [ 11 ]. V prvem primeru strokovnjaki opredeljujejo klasično lisencefalijo (tip I), v drugem pa – vezano na kromosom X. [ 12 ]
Ko se gen FLN1, ki kodira fosfoprotein filamin 1, izbriše, se proces usmerjene migracije nevronov morda sploh ne začne, kar vodi v popolno odsotnost zvitkov (agirija). [ 13 ]
Mutacije so bile ugotovljene v genu CDK5, ki kodira encim kinazo – katalizator za znotrajcelični metabolizem, ki uravnava celični cikel v nevronih centralnega živčnega sistema in zagotavlja njihovo normalno migracijo med prenatalnim nastajanjem možganskih struktur.
Nenormalne spremembe gena RELN na kromosomu 7, ki povzročajo kortikalne giralne defekte pri Norman-Robertsovem sindromu, povzročijo pomanjkanje zunajceličnega glikoproteina reelina, ki je potreben za uravnavanje migracije in pozicioniranja živčnih matičnih celic med razvojem možganske skorje.[ 14 ], [ 15 ], [ 16 ]
Gen ARX kodira nearistalens homeobox protein, transkripcijski faktor, ki igra pomembno vlogo v sprednjem delu možganov in drugih tkivih.[ 17 ] Otroci z mutacijo ARX imajo druge simptome, kot so manjkajoči deli možganov (ageneza corpus callosum), nenormalni genitalije in huda epilepsija.[ 18 ],[ 19 ]
Z lisencefalijo je bilo povezanih več genov. Med te gene spadajo VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A in CDK5.[ 20 ]
Citomegalovirus (CMV) je bil povezan z razvojem lisencefalije zaradi zmanjšane prekrvavitve možganov ploda. Resnost okužbe s CMV je odvisna od gestacijske starosti. Zgodnja okužba pogosteje povzroči lisencefalijo, ker se migracija nevronov pojavi zgodaj v nosečnosti.[ 21 ]
Poleg tega mehanizem nastanka te anomalije vključuje nepopolno ali poznejšo prekinitev migracije nevronov iz periventrikularne generativne cone v možgansko skorjo. V takih primerih se razvije bodisi nepopolna lisencefalija bodisi pahigirija, pri kateri se oblikuje več širokih žlebov in zvitkov (vendar jih večina ni).
Simptomi lissencefalija
Prvi znaki te patologije (če ni prej omenjenih sindromov) se morda ne pojavijo takoj po rojstvu, temveč po enem in pol do dveh mesecih. Najpogosteje pa opazimo naslednje klinične simptome lisencefalije:
- mišična hipotonija, pogosto v kombinaciji s spastično paralizo;
- konvulzije in generalizirani tonično-klonični napadi (v obliki opistotonusa);
- huda duševna zaostalost in zaostanek v rasti;
- okvara nevroloških in motoričnih funkcij.
Težave s požiranjem povzročajo težave pri hranjenju dojenčka. [ 22 ]
Visoka stopnja nevromotorične okvare se pogosto kaže kot tetraplegija – paraliza vseh okončin. Možna je deformacija rok, prstov na rokah ali nogah.
Pri Norman-Robertsovem sindromu z lisencefalijo tipa I opazimo kraniofacialne anomalije: hudo mikrocefalijo, nizek naklon čela in štrleč širok nosni most, široko postavljene oči (hiperterlorizem), nerazvitost čeljusti (mikrognatija). [ 23 ]
Za Millerjev-Diekerjev sindrom je lahko značilna tudi nenormalno majhna glava s širokim, visokim čelom in kratkim nosom, vdolbine v sencih (bitemporalne vdolbine) in nizko nastavljena, deformirana ušesa.
Za hud sindrom lisencefalije je značilna mikrocefalija, zmanjšanje velikosti zrkel (mikroftalmija), v kombinaciji z displazijo mrežnice, obstruktivnim hidrocefalusom in odsotnim ali hipoplastičnim corpus callosumom.
Zapleti in posledice
Med zapleti te anomalije strokovnjaki omenjajo motnje požiranja (disfagija) in gastroezofagealni refluks; refraktorno (nenadzorovano) epilepsijo; pogoste okužbe zgornjih dihal; pljučnico (vključno s kronično aspiracijo).
Dojenčki z lisencefalijo imajo lahko prirojene organske srčne težave v obliki defekta atrijskega septuma ali kompleksne srčne napake s cianozo (tetralogija Fallot). [ 24 ]
Posledice postnatalnega zaostanka v rasti so v večini primerov smrt v 24 mesecih po rojstvu.
Diagnostika lissencefalija
Diagnoza se začne s fizičnim pregledom otroka, preučevanjem zdravstvene anamneze staršev ter anamneze nosečnosti in poroda.
Med nosečnostjo bo morda potrebno testiranje fetalne DNK brez celic, amniocenteza ali odvzem vzorca horionskih resic. [ 25 ] Za več informacij glejte – Prenatalna diagnostika prirojenih bolezni
Instrumentalna diagnostika se uporablja za vizualizacijo možganskih struktur in oceno njihovih funkcij:
- računalniška tomografija možganov;
- slikanje z magnetno resonanco (MRI) možganov;
- elektroencefalogram (EEG). [ 26 ]
Med nosečnostjo lahko na ultrazvoku ploda po 20-21 tednih posumimo na lisencefalijo, če ni prisotnih parieto-okcipitalnih in kalkarnih žlebov ter anomalije Sylvijske fisure možganov.
Diferencialna diagnoza
Izvaja se diferencialna diagnostika z drugimi sindromi prirojenih možganskih napak.
Obstaja več kot 20 vrst lisencefalije, večina jih spada v dve glavni kategoriji: klasična lisencefalija (tip 1) in tlakovana lisencefalija (tip 2). Vsaka kategorija ima podobne klinične manifestacije, vendar različne genetske mutacije.[ 27 ]
Pregled možganov pri lisencefaliji tipa I pokaže možgansko skorjo s štirimi plastmi namesto šestih, kot jih vidimo pri normalnih bolnikih, medtem ko je pri lisencefaliji tipa 2 možganska skorja neorganizirana in se zdi grudasta ali nodularna zaradi popolnega premestitve možganske skorje s skupki kortikalnih nevronov, ločenih z gliomezenhimskim tkivom. Bolniki so imeli tudi mišične in očesne nepravilnosti.
- Klasična lisencefalija (tip 1):
- LIS1: Izolirana lisencefalija in Miller-Diekerjev sindrom (lisencefalija, povezana z obrazno dismorfijo). [ 28 ]
- LISX1: mutacija gena DCX. V primerjavi z lisencefalijo, ki jo povzročajo mutacije LIS1, ima DCX šestplastno skorjo namesto štirih.
- Izolirana lisencefalija brez drugih znanih genetskih okvar
- Tlakovana lisencefalija (tip 2):
- Walker-Warburgov sindrom
- Fukuyamin sindrom
- Bolezni mišic, oči in možganov
- Drugih vrst ni mogoče uvrstiti v eno od zgornjih dveh skupin:
- LIS2: Norman-Robertsov sindrom, podoben lisencefaliji tipa I ali Miller-Diekerjevemu sindromu, vendar brez delecije kromosoma 17.
- LIS3
- LISX2
Mikrolisencefalija: To je kombinacija odsotnosti normalnega kortikalnega gubanja in nenormalno majhne glave. Otroci z običajno lisencefalijo imajo ob rojstvu normalno velikost glave. Otroci z majhno velikostjo glave ob rojstvu so običajno diagnosticirani z mikrolisencefalijo.
Pomembno je tudi razlikovati med lisencefalijo in polimikrogirijo, ki sta različni možganski razvojni napaki.
Koga se lahko obrnete?
Zdravljenje lissencefalija
Lisencefalija je neozdravljiva organska okvara, zato je možno le podporno in simptomatsko zdravljenje. [ 29 ]
Najprej gre za uporabo antikonvulzivov in antiepileptikov, pa tudi za namestitev gastrostomske cevi v želodec (če otrok ne more samostojno pogoltniti). Masaža je koristna.
V primerih hudega hidrocefalusa se odstrani cerebrospinalna tekočina.
Preprečevanje
Strokovnjaki priporočajo, da bodoči starši poiščejo genetsko svetovanje, nosečnice pa naj se pravočasno prijavijo pri porodničarjih in ginekologih ter opravijo vse načrtovane preglede.
Napoved
Pri otrocih z lisencefalijo je prognoza odvisna od njene stopnje, vendar najpogosteje otrokov duševni razvoj ne preseže ravni štirih do petih mesecev. In vsi otroci s to diagnozo trpijo zaradi hudih psihomotoričnih motenj in težko ozdravljive epilepsije. [ 30 ]
Po podatkih NINDS (Ameriškega nacionalnega inštituta za nevrološke motnje in možgansko kap) je najdaljša pričakovana življenjska doba ljudi z lisencefalijo približno 10 let.