
Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
Waldenströmov B-celični limfoplazmatični limfom
Medicinski strokovnjak članka

Zadnji pregled: 12.07.2025
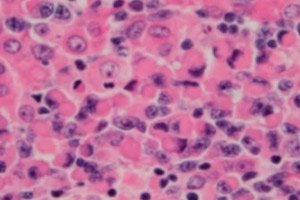
Maligna limfoproliferativna (imunoproliferativna) motnja, limfoplazmacitni limfom ali Waldenstromova makroglobulinemija, je celična neoplazma majhnih B-limfocitov – B-celic, ki zagotavljajo zaščitne funkcije limfnega sistema in humoralno imunost telesa. Diagnozo je treba postaviti šele po izključitvi vseh drugih majhnih B-celičnih limfomov. Waldenstromovo makroglobulinemijo je leta 1944 opisal Jan G. Waldenstrom, ki je pri dveh bolnikih poročal o nenavadnih manifestacijah limfadenopatske krvavitve, anemije, povečane sedimentacije, hiperviskoznosti in hipergamaglobulinemije. [ 1 ], [ 2 ]
Epidemiologija
Ta vrsta limfoma je redka, počasna hematološka maligna bolezen, klinična statistika pa ocenjuje stopnjo odkrivanja v tej skupini bolezni na približno 2 %. Poleg tega je moških bolnikov skoraj dvakrat več kot žensk.
Po nekaterih podatkih je incidenca letnih primerov limfoplazmocitnega limfoma v evropskih državah eden na 102 tisoč ljudi, v ZDA pa eden na 260 tisoč. [ 3 ]
Vzroki limfoplazmocitni limfom
Do danes etiologija večine onkoloških bolezni ostaja neznana, vendar se raziskave genetske osnove nekaterih od njih nadaljujejo. Raziskovalci so pri preučevanju vzrokov malignih bolezni plazemskih celic, vključno z limfoplazmocitnim limfomom B-celic – Waldenstromovo makroglobulinemijo, odkrili povezavo med patološko proliferacijo (delitvijo celic) limfocitov B v pozni fazi njihove diferenciacije in prisotnostjo določenih molekularno-genskih motenj, ki spreminjajo osnovne celične funkcije.
Pri bolnikih z Waldenstromovo makroglobulinemijo so bile ugotovljene spremembe v nekaterih genih - somatske mutacije, ki prizadenejo le tkiva s poškodbo genov ločene klonske populacije celic in tvorijo različice njihovega genoma, kar vodi do cikličnih in strukturnih motenj na celični ravni.
Najprej gre za somatske mutacije gena MYD88 (L265P) in CXCR4, ki kodira citosolni protein, pomemben za prirojeni in prilagodljivi imunski odziv: kot adapter zagotavlja prenos signalov iz provnetnega mediatorja IL-1 (interlevkin-1) in Toll-like receptorskih celic, ki aktivirajo imunski odziv. Zaradi somatske mutacije nastanejo anomalije v polipeptidni verigi molekule tega proteina – njegovi strukturni osnovi. [ 4 ]
Dejavniki tveganja
Poleg splošnih dejavnikov tveganja (izpostavljenost povišanim ravnem sevanja, rakotvornim kemikalijam itd.) veljajo za napovedovalce povečane verjetnosti razvoja Waldenstromove makroglobulinemije kot limfoproliferativne bolezni nizke stopnje naslednji dejavniki:
- starost (nad 65 let);
- prisotnost sorodnikov s to diagnozo, pa tudi z ne-Hodgkinovim limfomom B-celic ali kronično limfocitno levkemijo;
- kronični hepatitis C;
- anamneza benigne monoklonske gamopatije, idiopatske hematološke bolezni, katere bistvo je tvorba nenormalno spremenjenih gama globulinov tipa M s strani limfocitov plazemskih celic;
- avtoimunske bolezni, zlasti Sjögrenov sindrom.
Patogeneza
Ob stiku z antigenom ali stimulaciji s strani T-limfocitov se nekateri B-limfociti preoblikujejo v plazemske celice – limfocitne plazemske celice, ki po določenih transformacijah začnejo proizvajati zaščitne globularne beljakovine, torej gama globuline (imunoglobuline ali protitelesa).
Patogeneza limfoplazmocitnega limfoma/Waldenstromove makroglobulinemije vključuje hiperproliferacijo celic B, presežek klona limfocitnih plazemskih celic in prekomerno proizvodnjo imunoglobulina M (IgM), imenovanega tudi monoklonski imunoglobulin ali protein M, v krvi. To je glavno protitelo z veliko molekulsko maso in pentamerno strukturo, ki nastane med začetnim napadom na specifične bakterijske ali virusne antigene. [ 5 ]
Skoraj vsi simptomi te bolezni so povezani z manifestacijami aktivnosti M-proteina, ki lahko moti reološke lastnosti krvi, poveča njeno viskoznost; prodre v limfoidno in mieloidno tkivo kostnega mozga, se kopiči v perifernih limfoidnih tkivih (z nastankom počasi rastočih neoplazem, ki lahko pritiskajo na okoliške organe, živčna vlakna ali krvne žile).
Čeprav so kronična limfocitna levkemija, Waldenstromova makroglobulinemija ali limfoplazmacitni limfom in multipli mielom ločene bolezni, vse vključujejo povečano proliferacijo limfocitov B.
Simptomi limfoplazmocitni limfom
Prvi znaki bolezni so nespecifični in lahko vključujejo šibkost in povečano utrujenost (zaradi razvoja normokromne anemije), izgubo teže, težko dihanje, nočno hiperhidrozo in ponavljajočo se subfebrilno vročino.
Poleg tega se v začetni fazi bolezni pojavi motena občutljivost rok in nog, pojavi se periferna nevropatija (otrplost ali mravljinčenje v stopalih in nogah), pojavijo se majhne žariščne krvavitve kožnih kapilar (purpura) in hladna urtikarija (zaradi nastajanja in agregacije nenormalnih krioglobulinskih beljakovin v krvnem serumu).
Simptomi, povezani s sindromom hiperviskoznosti, vključujejo glavobole in omotico, poškodbe mrežnice in izgubo vida, tinitus in izgubo sluha, krče, bolečine v mišicah, visok krvni tlak, spontane krvavitve iz nosu in krvavitve dlesni. Pri ženskah se lahko pojavi krvavitev iz maternice.
Opažene so tudi: povečane bezgavke (limfadenopatija); povečana vranica (splenomegalija); srčno popuščanje s kardialgijo in motnjami srčnega ritma. Čeprav je visceralna infiltracija redka, sta lahko prizadeta želodec in črevesje, z razvojem driske (pogosto z mastnim blatom). [ 6 ], [ 7 ]
Obrazci
Klasifikacija tumorjev hematopoetskega in limfoidnega tkiva Svetovne zdravstvene organizacije iz leta 2017 določa štiri diagnostična merila za Waldenstromovo makroglobulinemijo, vključno z:
- Prisotnost monoklonske IgM gamopatije
- Infiltracija kostnega mozga z majhnimi limfociti, ki kažejo plazmacitoidno ali plazemskocelično diferenciacijo
- Infiltracija kostnega mozga z intertrabekularno strukturo
- Imunofenotip, skladen z Waldenstromovo makroglobulinemijo, ki vključuje površinske IgM+, CD19+, CD20+, CD22+, CD25+, CD27+, FMC7+, variabilne CD5, CD10-, CD23-, CD103- in CD108-
Zapleti in posledice
Bolniki z limfoplazmocitnim limfomom razvijejo zaplete in posledice v obliki:
- zmanjšana imunost;
- odpoved kostnega mozga z motnjami njegovih hematopoetskih funkcij in razvojem anemije;
- pomanjkanje oblikovanih elementov krvi, kot so eritrociti, levkociti, trombociti;
- lezije prebavil s kronično drisko in moteno črevesno absorpcijo (sindrom malabsorpcije);
- vnetje sten krvnih žil (kompleksni imunski vaskulitis);
- povečana krhkost kosti (osteoporoza);
- okvare vida in sluha;
- sekundarna amiloidoza notranjih organov;
- napredovanje v paraproteinemično hemoblastozo v obliki multiplega mieloma;
- transformacija v zelo maligni tip limfoma – difuzni velikocelični B-celični limfom.
Diagnostika limfoplazmocitni limfom
Diagnoza limfoplazmocitnega limfoma/Waldenstromove makroglobulinemije je običajno težka zaradi pomanjkanja specifičnih morfoloških, imunofenotipskih ali kromosomskih sprememb. Zaradi te pomanjkljivosti je razlikovanje te bolezni od drugih limfomov drobnih B-celic izključujoče.[ 8 ]
Poleg ocene obstoječih simptomov so za diagnozo limfoplazmocitnega limfoma potrebni splošni in biokemični krvni test, koagulogram, imunoelektroforeza krvnih beljakovin z določitvijo ravni imunoglobulina M v krvi in splošni test urina. [ 9 ]
Potrebna je biopsija kostnega mozga, za katero se opravi punkcija kostnega mozga.
Izvajajo se instrumentalne diagnostike: ultrazvok bezgavk in vranice, rentgensko slikanje kosti, CT prsnega koša in trebušne votline, oftalmoskopija.
Diferencialna diagnoza
Limfoplazmacitni limfom velja za diagnozo izključitve, zato se diferencialna diagnoza izvaja s kronično limfocitno levkemijo B-celic, multiplim mielomom, folikularnim limfomom, različnimi podtipi ne-Hodgkinovega limfoma, plazmocitomom, reaktivno plazmocitozo, angiofolikularno limfoidno hiperplazijo (Castlemanova bolezen) itd.
Koga se lahko obrnete?
Zdravljenje limfoplazmocitni limfom
Upoštevati je treba, da je Waldenstromova makroglobulinemija ali limfoplazmocitni limfom lahko asimptomatska več let in se diagnosticira s povečano raven M-proteina v krvi.
Če ni simptomov, se izvaja aktivno spremljanje z rednimi pregledi in testi.
Na podlagi obstoječih simptomov in rezultatov laboratorijskih preiskav se sprejme odločitev o začetku terapije, ki je odvisna od številnih dejavnikov (npr. starosti, napredovanja bolezni itd.).
V skladu s protokolom je začetno zdravljenje bolnikov s to vrsto limfoma običajno kombinacija radioterapije in kemoterapije z dajanjem citostatikov, kot so ciklofosfamid, doksorubicin, vinkristin, ter kortikosteroidov - metprednizolona ali deksametazona.
Učinkovitost kemoterapije z zdravili iz skupine monoklonskih protiteles, zlasti z rituksimabom, je bila dokazana. [ 10 ]
V primerih generalizirane bolezni se Rituksimab uporablja v kombinaciji z protitumorskimi nukleozidnimi analogi (pentostatin, kladribin). Pri počasi napredujoči bolezni z nizkimi ravnmi monoklonskega imunoglobulina M se poleg Rituksimaba uporablja tudi citostatik Klorambucil (Leukeran). [ 11 ]
Za zmanjšanje viskoznosti krvi in stabilizacijo ravni njenih oblikovanih elementov se uporablja terapevtska hemafereza.
Ko je raven protiteles v krvi kritično nizka, se izvaja nadomestno zdravljenje z imunoglobulini, da se preprečijo sočasne ponavljajoče se okužbe.
Kot ugotavljajo onkohematologi, kljub temu, da lahko zdravljenje privede do remisije bolezni, večina bolnikov doživi ponovitev bolezni. Če se pojavi prej kot v 24 mesecih, se lahko uporabi protitumorsko zdravilo, kot je Ibrutinib (v obliki tablet). V primeru kasnejših ponovitev se zdravljenje izvaja po prvotni shemi. [ 12 ], [ 13 ], [ 14 ]
Preprečevanje
Strokovnjaki določajo prognozo izida limfoplazmocitnega limfoma v skladu z mednarodnim prognostičnim sistemom za ocenjevanje glavnih parametrov: bolnikove starosti in serumskih ravni hemoglobina, trombocitov, beta-2-mikroglobulina in monoklonskega imunoglobulina. [ 15 ], [ 16 ]
Povprečna stopnja preživetja pri tej diagnozi je približno pet let, vendar skoraj 40 % bolnikov živi deset let ali več.