
Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
Achondroplazija
Medicinski strokovnjak članka

Zadnji pregled: 12.07.2025
Obstaja veliko redkih prirojenih bolezni, ena od njih pa je kršitev rasti kosti - ahondroplazija, ki vodi do hude nesorazmerne nizke rasti.
V poglavju o razvojnih anomalijah ICD-10 je koda za to vrsto dedne osteohondralne displazije z rastnimi napakami cevastih kosti in hrbtenice Q77.4 [ 1 ]
Epidemiologija
Glede razširjenosti ahondroplazije so statistični podatki različnih študij dvoumni. Nekateri trdijo, da se ta anomalija pojavi pri enem novorojenčku od 10 tisoč, drugi - pri enem od 26-28 tisoč, tretji pa - pri 4-15 primerih od 100 tisoč. [ 2 ]
Obstajajo tudi podatki, da je incidenca ahondroplazije pri otrocih, ko je oče starejši od 50 let, en primer na 1875 novorojenčkov.
Vzroki ahondroplazija
Vzrok ahondroplazije je kršitev osteogeneze, zlasti ena od vrst intrauterine osifikacije diafiz cevastih kosti okostja - endohondralna osifikacija, med katero se hrustanec spremeni v kostno tkivo. Za več podrobnosti glejte - Razvoj in rast kosti
Motnje osifikacije dolgih kosti, tj. fetalna ahondroplazija, nastanejo zaradi mutacij v genu membranske tirozin kinaze - receptorja 3 za rastni faktor fibroblastov (FGFR3 na kromosomu 4p16.3), ki vpliva na rast in diferenciacijo celic. Prisotnost mutacij FGFR3 je povezana z genetsko nestabilnostjo in spremembami v številu kromosomov (anevploidija).
Ahondroplazija se na otroka prenaša kot avtosomno dominantna lastnost, torej prejme eno kopijo mutantnega gena (ki je dominanten) in en normalen gen na paru nespolnih (avtosomnih) kromosomov. Torej je tip dedovanja te napake avtosomno dominanten, anomalija pa se lahko pojavi pri 50 % potomcev, če se križa kombinacija alelov tega gena (genotip).
Poleg tega so mutacije lahko sporadične in, kot kaže praksa, se v 80 % primerov otroci z ahondroplazijo rodijo staršem normalne višine.
Dejavniki tveganja
Glavni dejavniki tveganja za rojstvo otrok z ahondroplazijo so dedni. Če ima eden od staršev to napako, je verjetnost rojstva bolnega otroka ocenjena na 50 %; če imata oba starša to anomalijo, je tudi 50 %, vendar s 25 % tveganjem za homozigotno ahondroplazijo, ki vodi v smrt pred rojstvom ali v zgodnjem povojnem obdobju.
S starostjo očeta (bližje 40. letu in več) se povečuje tveganje za novo mutacijo (de novo mutacijo) gena FGFR3.
Patogeneza
Strokovnjaki pri razlagi patogeneze ahondroplazije poudarjajo pomen transmembranske proteinske tirozin proteinske kinaze (ki jo kodira gen FGFR3) pri uravnavanju delitve, diferenciacije in apoptoze celic hrustančnega tkiva rastnih plošč - hondrocitov, pa tudi pri normalnem razvoju okostja - osteogenezi in mineralizaciji kostnega tkiva.
Med embrionalnim razvojem, ob prisotnosti genske mutacije, postanejo receptorji fibroblastnega rastnega faktorja 3 bolj aktivni. Povečanje njihovih funkcij moti prenos celičnih signalov in interakcijo zunajceličnega dela tega proteina s polipeptidnimi fibroblastnimi rastnimi faktorji (FGF). Posledično pride do okvare: faza proliferacije hrustančnih celic se skrajša, njihova diferenciacija pa se začne prej, kot je bilo pričakovano. Vse to vodi do nepravilnega nastajanja in zlivanja lobanjskih kosti ter skeletne displazije - zmanjšanja dolgih kosti, kar spremlja izrazita nizka rast ali pritlikavost.
In dve tretjini primerov pritlikavosti sta povezani z ahondroplazijo.
Simptomi ahondroplazija
Nenormalna rast kosti povzroča klinične simptome ahondroplazije, kot so:
- izrazita nizka rast (nesorazmeren pritlikavost) s povprečno odraslo višino 123-134 cm;
- skrajšanje proksimalnih delov spodnjih in zgornjih okončin ob relativno normalni velikosti trupa;
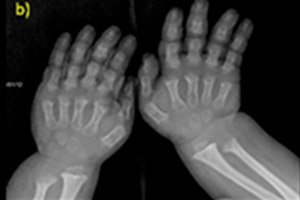
- skrajšani prsti na rokah in nogah;
- povečana glava (makro ali megalocefalija); [ 3 ]
- specifične obrazne poteze v obliki štrlečega čela in hipoplazije srednjega dela obraza - vdrt nosni most.
- ozek kraniocervikalni prehod. Nekateri dojenčki z ahondroplazijo umrejo v prvem letu življenja zaradi zapletov, povezanih s kraniocervikalnim prehodom; populacijske študije kažejo, da je to povečano tveganje smrti lahko kar 7,5 % brez ocene in intervencije.[ 4 ]
- Disfunkcija srednjega ušesa je pogosto težava [ 5 ] in če se ne zdravi pravilno, lahko povzroči prevodno izgubo sluha, ki je dovolj huda, da ovira razvoj govora. Več kot polovica otrok bo potrebovala cev za izenačevanje tlaka. [ 6 ] Na splošno ima približno 40 % ljudi z ahondroplazijo funkcionalno pomembno izgubo sluha. Pogosto je tudi razvoj izraznega jezika zakasnjen, čeprav je moč povezave med izgubo sluha in težavami z izraznim jezikom vprašljiva.
- Ukrivljenost golenice je zelo pogosta pri ljudeh z ahondroplazijo. Več kot 90 % nezdravljenih odraslih ima določeno stopnjo ukrivljenosti.[ 7 ] "Ukrivljenost" je pravzaprav kompleksna deformacija, ki je posledica kombinacije bočnega nagiba, notranje torzije golenice in dinamične nestabilnosti kolena.[ 8 ]
Za dojenčke z ahondroplazijo je značilna mišična hipotonija, zaradi katere se gibalnih veščin in hodi začnejo učiti kasneje. Inteligenca in kognitivne sposobnosti zaradi te razvojne napake niso prizadete. [ 9 ], [ 10 ]
Posledice in zapleti
Za to vrsto dedne osteohondralne displazije so značilni naslednji zapleti in posledice:
- ponavljajoče se okužbe ušes;
- obstruktivna apneja v spanju;
- hidrocefalus;
- malokluzija in krivi zobje:
- deformacija nog (varus ali valgus) s spremembo hoje;
- hipertrofirana lordoza ledvene hrbtenice ali njena ukrivljenost (torakolumbalna kifoza ali ledvena skolioza) - z bolečinami v hrbtu pri hoji;
- bolečine v sklepih (zaradi nepravilne lege kosti ali stiskanja živčnih korenin);
- Spinalna stenoza in kompresija hrbtenjače; Najpogostejša zdravstvena težava v odrasli dobi je simptomatska spinalna stenoza, ki zajema L1-L4. Simptomi segajo od občasne, reverzibilne klavdikacije, ki jo povzroči vadba, do hude, ireverzibilne disfunkcije nog in zastajanja urina.[ 11 ] Klavdikacija in stenoza lahko povzročita tako senzorične (otrplost, bolečina, teža) kot motorične simptome (šibkost, spotikanje, omejena vzdržljivost pri hoji). Vaskularna klavdikacija je posledica otekanja krvnih žil po stanju in hoji ter je popolnoma reverzibilna s počitkom. Spinalna stenoza je dejanska lezija hrbtenjače ali živčne korenine s stenotično kostjo hrbtenjačnega kanala, simptomi pa so ireverzibilni. Simptomi, lokalizirani na določenem dermatomu, so lahko posledica stenoze specifičnih odprtin živčne korenine.
- zmanjšanje prsne stene z omejeno rastjo pljuč in zmanjšano pljučno funkcijo (huda zasoplost). V otroštvu ima majhna skupina ljudi z ahondroplazijo restriktivne težave s pljuči. Majhne prsi in povečana kompliancija prsnega koša skupaj povzročita zmanjšano pljučno kapaciteto in restriktivno pljučno bolezen [ 12 ].
Druge ortopedske težave
- Šibkost sklepov. Večina sklepov je v otroštvu hipermobilnih. Na splošno to nima velikega učinka, razen nestabilnosti kolena pri nekaterih ljudeh.
- Diskoidni lateralni meniskus: Ta nedavno odkrita strukturna anomalija lahko pri nekaterih ljudeh povzroči kronično bolečino v kolenu.[ 13 ]
- Artritis: Konstitutivna aktivacija FGFR-3, kot pri ahondroplaziji, lahko ščiti pred razvojem artritisa.[ 14 ]
- Črna akantoza se pojavlja pri približno 10 % ljudi z ahondroplazijo.[ 15 ] Pri tej populaciji ne odraža hiperinzulinemije ali malignosti.
Homozigotna ahondroplazija, ki jo povzročajo bialelne patogene variante na nukleotidu 1138 FGFR3, je huda motnja z radiološkimi izsledki, ki se kvalitativno razlikujejo od tistih pri ahondroplaziji. Zgodnja smrt je posledica odpovedi dihanja zaradi majhne prsne stene in nevroloških izpadov zaradi cervikomedularne stenoze [Hall 1988].
Diagnostika ahondroplazija
Pri večini bolnikov se diagnoza ahondroplazije postavi na podlagi značilnih kliničnih znakov in radiografskih izvidov. Pri dojenčkih ali v odsotnosti nekaterih simptomov se za dokončno diagnozo uporabi genetsko testiranje, kot je analiza kariotipa.[16 ]
Pri izvajanju prenatalne diagnostike z metodo molekularne genetike se lahko opravijo analize amnijske tekočine ali vzorca horionskih resic.
Znaki ahondroplazije na ultrazvoku ploda - skrajšanje okončin in tipične obrazne poteze - se vizualizirajo po 22. tednu nosečnosti.
Instrumentalna diagnostika vključuje tudi rentgensko slikanje okostja ali ultrazvok kosti. Rentgensko slikanje potrdi diagnozo na podlagi podatkov, kot so velika lobanja z ozkim okcipitalnim foramenom in relativno majhno bazo; kratke cevaste kosti in skrajšana rebra; kratka in sploščena vretenca; zožen hrbtenični kanal, zmanjšana velikost iliakalnih kril.
Diferencialna diagnoza
Diferencialna diagnoza s pritlikavostjo hipofize, prirojeno spondiloepifizno in diastrofno displazijo, hipohondroplazijo, Shereshevsky-Turnerjevim in Noonanovim sindromom ter psevdoahondroplazijo je torej normalna. Razlika med psevdoahondroplazijo in ahondroplazijo je torej v tem, da so pri bolnikih s pritlikavostjo pri psevdoahondroplaziji velikost glave in obrazne poteze normalne.
Koga se lahko obrnete?
Zdravljenje ahondroplazija
Priporočila za oskrbo otrok z ahondroplazijo je opisal Odbor za genetiko Ameriške akademije za pediatrijo. Ta priporočila so namenjena zagotavljanju smernic in ne nadomeščajo individualnega odločanja. Nedavni pregled [Pauli & Botto 2020] vključuje tudi smernice. Obstajajo specializirane klinike, ki so specializirane za zdravljenje skeletne displazije; njihova priporočila se lahko nekoliko razlikujejo od teh splošnih priporočil.
Priporočila vključujejo (vendar niso omejena na) naslednje.
Hidrocefalus. Če se pojavijo znaki ali simptomi povečanega intrakranialnega tlaka (npr. pospešena rast glave, vztrajno izbočena fontanela, opazna razširitev površinskih ven na obrazu, razdražljivost, bruhanje, spremembe vida, glavobol), je potrebna napotitev k nevrokirurgu.
Domnevna etiologija hidrocefalusa pri ahondroplaziji je povečan intrakranialni venski tlak zaradi stenoze jugularnih foramen. Zato je standardno zdravljenje ventrikuloperitonealna premestitev. Vendar pa je lahko endoskopska tretja ventrikulostomija koristna pri nekaterih posameznikih,[ 17 ] kar pomeni, da so lahko vključeni tudi drugi mehanizmi, kot je obstrukcija izhoda četrtega prekata zaradi kraniocervikalne stenoze.[ 18 ]
Stenoza kraniocervikalnega prehoda. Najboljši napovedovalci potrebe po subokcipitalni dekompresiji:
- Hiperrefleksija ali klonus spodnjih okončin
- Centralna hipopneja na polisomnografiji
- Zmanjšanje velikosti foramen magnuma, določeno z računalniško tomografijo kraniocervikalnega prehoda in primerjano z normami pri otrocih z ahondroplazijo.[ 19 ]
- Kot dodaten dejavnik, ki ga je treba upoštevati pri odločitvi za operacijo, so bili nedavno predlagani dokazi o kompresiji hrbtenjače in/ali nepravilnosti signala, uteženega na T2.
Če obstajajo jasni znaki simptomatske kompresije, je treba nujno napotiti otroka k pediatričnemu nevrokirurgu za dekompresijsko operacijo. [ 20 ]
Zdravljenje obstruktivne spalne apneje lahko vključuje:
- Adenotonzilektomija
- Pozitiven tlak v dihalnih poteh
- Traheostomija v skrajnih primerih
- Izguba teže
Ti posegi lahko izboljšajo motnje spanja in nekoliko izboljšajo nevrološko delovanje.[ 21 ]
V redkih primerih, ko je obstrukcija dovolj huda, da zahteva traheostomijo, se za lajšanje obstrukcije zgornjih dihalnih poti uporablja operacija napredovanja na srednji del obraza.[ 22 ]
Disfunkcija srednjega ušesa. Pogoste okužbe srednjega ušesa, vztrajno kopičenje tekočine v srednjem ušesu in posledično izgubo sluha je treba po potrebi agresivno zdraviti. Dolgotrajna uporaba cevk je priporočljiva, ker je pogosto potrebna do sedmega ali osmega leta starosti.[ 23 ]
Kadar se težave pojavijo v kateri koli starosti, je priporočljivo uporabiti ustrezne metode zdravljenja.
Nizka rast. Več študij je ocenilo terapijo z rastnim hormonom (GH) kot možno zdravljenje za ahondroplazijo nizke rasti.[ 24 ]
Na splošno te in druge serije kažejo začetno pospešitev rasti, vendar se učinek sčasoma zmanjšuje.
V povprečju lahko pričakujete povečanje odrasle višine le za približno 3 cm.
Podaljšanje okončin z uporabo različnih tehnik ostaja za nekatere možnost. Doseči je mogoče povečanje višine do 30–35 cm. [ 25 ] Zapleti so pogosti in lahko resni.
Medtem ko nekateri zagovarjajo izvajanje teh posegov že pri šestih do osmih letih, mnogi pediatri, klinični genetiki in etiki zagovarjajo odložitev takšne operacije, dokler mlada oseba ne bo sposobna sodelovati pri sprejemanju premišljene odločitve.
Vsaj v Severni Ameriki se le majhen delež prizadetih posameznikov odloči za podaljševanje okončin. Zdravstveni svetovalni odbor Little People of America je izdal izjavo glede uporabe podaljševanja okončin.
Debelost: Ukrepi za preprečevanje debelosti se morajo začeti v zgodnjem otroštvu. Standardno zdravljenje debelosti bi moralo biti učinkovito pri ljudeh z ahondroplazijo, čeprav so potrebe po kalorijah nižje. [ 26 ]
Za spremljanje napredka je treba uporabljati standardne tabele teže in razmerja med težo in višino, specifične za ahondroplazijo. Pomembno je omeniti, da te krivulje niso popolne krivulje razmerja med težo in višino; izpeljane so bile iz tisočih podatkovnih točk ljudi z ahondroplazijo.
Standardi indeksa telesne mase (ITM) so bili razviti za otroke, stare 16 let in manj. [ 27 ] ITM ni standardiziran za odrasle z ahondroplazijo; primerjave s krivuljami ITM za povprečno višino bodo dale zavajajoče rezultate. [ 28 ]
Varusna deformacija. Priporočljivo je letno ortopedsko spremljanje pri zdravniku, ki pozna ahondroplazijo, ali pri ortopedskem kirurgu. Objavljena so bila merila za kirurški poseg.[ 29 ]
Prisotnost progresivne simptomatske krivulje zahteva napotitev k ortopedu. Asimptomatska varusna deformacija sama po sebi običajno ne zahteva kirurške korekcije. Izbirate lahko med različnimi posegi (npr. vodena rast z osmimi ploščicami, valgusna osteotomija in derotacijska osteotomija). Ni kontroliranih študij, ki bi primerjale rezultate možnosti zdravljenja.
Kifoza. Dojenčki z ahondroplazijo pogosto razvijejo fleksibilno kifozo. Na voljo je protokol za preprečevanje razvoja fiksne kotne kifoze, ki vključuje izogibanje fleksibilnim vozičkom, gugalnicam in nosilkam za dojenčke. Nasvet proti sedenju brez podpore; pri držanju dojenčka vedno izvajajte protipritisk na hrbet.
- Kifoza se pri večini otrok znatno izboljša ali izgine po tem, ko zavzamejo ortogradno držo in začnejo hoditi. [ 30 ]
- Pri otrocih, ki se po povečanju moči trupa in začetku hoje spontano ne izboljšajo, je običajno zadostna opornica, da se prepreči vztrajanje torakolumbalne kifoze.[ 31 ]
- Če huda kifoza vztraja, bo morda potrebna operacija hrbtenice, da se preprečijo nevrološki zapleti.[ 32 ]
Spinalna stenoza: Če se pojavijo hudi znaki in/ali simptomi spinalne stenoze, je potrebna nujna napotitev k kirurškemu specialistu.
Običajno se priporoča razširjena in široka laminektomija. Pomembnost posega je odvisna od ravni (npr. prsni ali ledveni del) in stopnje stenoze. Bolniki so imeli boljše rezultate in izboljšano funkcijo prej, ko so imeli operacijo po pojavu simptomov [ 33 ].
Cepljenja: Nič v zvezi z ahondroplazijo ne izključuje vseh rutinskih cepljenj. Glede na povečano tveganje za okužbe dihal so še posebej pomembna cepiva proti DTaP, pnevmokoknim okužbam in gripi.
Prilagoditvene potrebe: Zaradi nizke rasti bodo morda potrebne prilagoditve okolja. V šoli so to lahko stoli, znižana stikala za luči, stranišča ustrezne višine ali druga sredstva za dostopnost, nižje mize in nasloni za noge pred stoli. Vsi otroci bi morali biti sposobni samostojno zapustiti stavbo v nujnih primerih. Majhne roke in šibke kite lahko otežijo finomotoriko. Ustrezne prilagoditve vključujejo uporabo manjše tipkovnice, obteženih pisal in bolj gladkih površin za pisanje. Večina otrok bi morala imeti individualiziran izobraževalni program (IEP) ali načrt 504.
Za vožnjo so skoraj vedno potrebni podaljški pedalov. Morda bodo potrebne tudi prilagoditve delovne postaje, kot so nižje mize, manjše tipkovnice, stopnice in dostop do stranišča.
Socializacija: Zaradi zelo opazne nizke rasti, povezane z ahondroplazijo, imajo lahko prizadeti posamezniki in njihove družine težave s socializacijo in prilagajanjem na šolo.
Podporne skupine, kot je Little People of America, Inc (LPA), lahko družinam pomagajo pri reševanju teh težav s pomočjo vrstniške podpore, osebnega zgleda in programov socialnega ozaveščanja.
Informacije o zaposlovanju, izobraževanju, pravicah invalidov, posvojitvi otrok nizke rasti, zdravstvenih težavah, ustreznih oblačilih, prilagodljivih pripomočkih in starševstvu so na voljo v nacionalnem glasilu, na seminarjih in delavnicah.
Ni zdravila ali zdravljenja brez zdravil, ki bi lahko pozdravilo to prirojeno napako.
Najpogosteje se uporablja fizioterapija; zdravljenje je lahko potrebno tudi pri hidrocefalusu (s šantom ali endoskopsko ventrikulostomijo), debelosti, [ 34 ] apneji, [ 35 ] okužbi srednjega ušesa ali spinalni stenozi.
V nekaterih klinikah po otrokovem petem do sedmem letu starosti izvajajo kirurško zdravljenje: podaljševanje kosti goleni, stegen in celo ramenskih kosti ali korekcijo deformacije - s pomočjo operacij in posebnih ortopedskih pripomočkov - v treh do štirih fazah, vsaka traja do 6-12 mesecev.
Terapija v preiskavi
Uporaba analoga natriuretičnega peptida tipa C je v kliničnih preskušanjih. Začetni rezultati so pokazali, da ga otroci z ahondroplazijo dobro prenašajo in da pri otrocih z ahondroplazijo ( mesto preskušanja ) povzroči povečanje hitrosti rasti od izhodišča. [ 36 ] Konjugirani natriuretični peptid tipa C je trenutno prav tako v kliničnih preskušanjih. [ 37 ] Drugi dejavniki vključujejo zaviranje tirozin kinaze [ 38 ], meklizin [ 39 ] in topno rekombinantno humano vabo FGFR3. [ 40 ]
Za informacije o kliničnih preskušanjih za širok spekter bolezni in stanj poiščite spletno stran clinicaltrials.gov v ZDA in register kliničnih preskušanj EU v Evropi.
Preprečevanje
Edini preventivni ukrep je prenatalna diagnoza prirojenih bolezni. [ 41 ], [ 42 ]
Napoved
Kako dolgo živijo ljudje z ahondroplazijo? Približno 10 let manj od povprečne pričakovane življenjske dobe.
Ker patološke spremembe v kostnem tkivu in sklepih vodijo do omejitev samooskrbe in gibljivosti, se otrokom s to diagnozo podeli status invalida. Dolgoročno ima večina bolnikov normalno prognozo, vendar se s starostjo povečuje tveganje za srčne bolezni. [ 43 ]