
Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
Dedni nefritis (Alportov sindrom) pri otrocih
Medicinski strokovnjak članka

Zadnji pregled: 05.07.2025
Dedni nefritis (Alportov sindrom) je genetsko določena dedna neimunska glomerulopatija, ki se kaže s hematurijo (včasih s proteinurijo), progresivnim upadanjem delovanja ledvic z razvojem kronične ledvične odpovedi, pogosto v kombinaciji s senzorinevralno gluhostjo in okvaro vida.
Bolezen je leta 1902 prvič opisal LG Guthrie, ki je opazoval družino, v kateri so hematurijo opazovali v več generacijah. Leta 1915 je A. F. Hurst opisal razvoj uremije pri članih iste družine. Leta 1927 je A. Alport prvi ugotovil izgubo sluha pri več sorodnikih s hematurijo. V petdesetih letih prejšnjega stoletja so opisali očesne lezije pri podobni bolezni. Leta 1972 so Hinglais in sodelavci pri bolnikih z dedno hematurijo med morfološko študijo ledvičnega tkiva odkrili neenakomerno širitev in stratifikacijo glomerularnih bazalnih membran. Leta 1985 so identificirali genetsko osnovo dednega nefritisa - mutacijo v genu za kolagen tipa IV (Fiengold in sodelavci, 1985).
Študija genetske narave bolezni nam je omogočila sklep, da so razlike v fenotipskih manifestacijah dednega nefritisa (z izgubo sluha ali brez nje) posledica stopnje izražanja mutantnega gena. Zato se trenutno vse klinične različice obravnavajo kot manifestacije ene bolezni, izraz "dedni nefritis" pa je sinonim za izraz "Alportov sindrom".
Glede na epidemiološke študije se dedni nefritis pojavlja s pogostostjo 17 na 100.000 otrok.
 [
[ Vzroki Alportovega sindroma
Genetska osnova bolezni je mutacija v genu verige a-5 kolagena tipa IV. Ta tip je univerzalen za bazalne membrane ledvic, kohlearni aparat, lečno kapsulo, mrežnico in roženico očesa, kar je bilo dokazano v študijah z uporabo monoklonskih protiteles proti tej frakciji kolagena. V zadnjem času se kaže možnost uporabe DNK sond za prenatalno diagnostiko dednega nefritisa.
Poudarjen je pomen testiranja vseh družinskih članov z DNK sondami za identifikacijo nosilcev mutantnega gena, kar je zelo pomembno pri izvajanju medicinsko-genetskega svetovanja družin s to boleznijo. Vendar pa do 20 % družin nima sorodnikov, ki trpijo za boleznijo ledvic, kar kaže na visoko pogostost spontanih mutacij nenormalnega gena. Večina bolnikov z dednim nefritisom ima v svojih družinah posameznike z boleznijo ledvic, izgubo sluha in patologijo vida; pomembne so krvne poroke med ljudmi z enim ali več predniki, saj se v zakonski zvezi sorodnih posameznikov poveča verjetnost prejema istih genov od obeh staršev. Ugotovljene so bile avtosomno dominantne, avtosomno recesivne in dominantne, na X vezane poti prenosa.
Pri otrocih najpogosteje ločimo tri vrste dednega nefritisa: Alportov sindrom, dedni nefritis brez izgube sluha in družinska benigna hematurija.
Alportov sindrom je dedni nefritis z okvaro sluha. Temelji na kombinirani okvari strukture kolagena glomerularne bazalne membrane ledvic, ušesnih in očesnih struktur. Gen klasičnega Alportovega sindroma se nahaja v lokusu 21-22 q dolgega kraka kromosoma X. V večini primerov se deduje dominantno, povezano s kromosomom X. V zvezi s tem je Alportov sindrom pri moških hujši, saj pri ženskah funkcijo mutantnega gena kompenzira zdrav alel drugega, nepoškodovanega kromosoma.
Genetska osnova za razvoj dednega nefritisa so mutacije v genih alfa verig kolagena tipa IV. Znanih je šest alfa verig kolagena tipa IV G: geni verig a5 in a6 (Col4A5 in Col4A5) se nahajajo na dolgem kraku kromosoma X v coni 21-22q; geni verig a3 in a4 (Col4A3 in Col4A4) so na 2. kromosomu; geni verig a1 in a2 (Col4A1 in Col4A2) so na 13. kromosomu.
V večini primerov (80–85 %) se odkrije na X vezan vzorec dedovanja bolezni, povezan s poškodbo gena Col4A5 zaradi delecije, točkovnih mutacij ali motenj spajanja. Trenutno je bilo odkritih več kot 200 mutacij gena Col4A5, ki so odgovorne za motnjo sinteze a5-verig kolagena tipa IV. Pri tej vrsti dedovanja se bolezen kaže pri otrocih obeh spolov, vendar je pri dečkih hujša.
Mutacije v lokusih genov Col4A3 in Col4A4, odgovornih za sintezo verig a3 in a4 kolagena tipa IV, se dedujejo avtosomno. Glede na raziskave je avtosomno dominantni tip dedovanja opažen pri 16 % primerov dednega nefritisa, avtosomno recesivni tip pa pri 6 % bolnikov. Znanih je približno 10 variant mutacij genov Col4A3 in Col4A4.
Posledica mutacij je kršitev procesov sestavljanja kolagena tipa IV, kar vodi v kršitev njegove strukture. Kolagen tipa IV je ena glavnih komponent glomerularne bazalne membrane, kohlearnega aparata in očesne leče, katere patologija bo odkrita v kliniki dednega nefritisa.
Kolagen tipa IV, ki je del glomerularne bazalne membrane, je sestavljen predvsem iz dveh verig a1 (IV) in ene verige a2 (IV), vsebuje pa tudi verige a3, a4 in a5. Najpogosteje pri dedovanju, vezanem na X, mutacijo gena Col4A5 spremlja odsotnost verig a3, a4, a5 in a6 v strukturi kolagena tipa IV, število verig o1 in a2 v glomerularni bazalni membrani pa se poveča. Mehanizem tega pojava ni jasen, domneva se, da so vzrok posttranskripcijske spremembe mRNA.
Odsotnost verig a3, a4 in a5 v strukturi kolagena tipa IV glomerularnih bazalnih membran vodi v njihovo tanjšanje in krhkost v zgodnjih fazah Alportovega sindroma, kar se klinično pogosteje kaže s hematurijo (redkeje s hematurijo s proteinurijo ali samo s proteinurijo), izgubo sluha in lentikonusom. Nadaljnje napredovanje bolezni vodi v odebelitev in oslabljeno prepustnost bazalnih membran v poznih fazah bolezni s proliferacijo kolagena tipov V in VI v njih, kar se kaže v povečani proteinuriji in zmanjšani ledvični funkciji.
Narava mutacije, ki je osnova dednega nefritisa, v veliki meri določa njegovo fenotipsko manifestacijo. V primeru delecije kromosoma X s sočasno mutacijo genov Col4A5 in Col4A6, odgovornih za sintezo a5- in a6-verig kolagena tipa IV, se Alportov sindrom kombinira z leiomiomatozo požiralnika in genitalij. Glede na raziskovalne podatke je v primeru mutacije gena Col4A5, povezane z delecijo, opažena večja resnost patološkega procesa, kombinacija ledvične okvare z zunajledvičnimi manifestacijami in zgodnji razvoj kronične ledvične odpovedi v primerjavi s točkovno mutacijo tega gena.
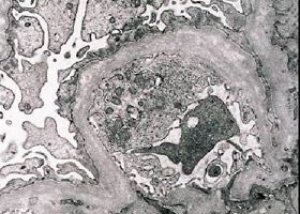
Morfološko elektronska mikroskopija razkriva stanjšanje in stratifikacijo glomerularnih bazalnih membran (zlasti lamina densa) ter prisotnost elektronsko gostih granul. Glomerularne lezije so lahko pri istem bolniku heterogene, od minimalnih fokalnih mezangialnih lezij do glomeruloskleroze. Glomerulitis pri Alportovem sindromu je vedno imunonegativen, kar ga loči od glomerulonefritisa. Značilne značilnosti vključujejo razvoj tubularne atrofije, limfohistiocitno infiltracijo in prisotnost "penastih celic" z lipidnimi vključki - lipofagi. Z napredovanjem bolezni se razkrijeta odebelitev in izrazito uničenje glomerularnih bazalnih membran.
Razkrijejo se določene spremembe v imunskem sistemu. Bolniki z dednim nefritisom imajo znižano raven Ig A in nagnjenost k zvišani koncentraciji IgM v krvi, raven IgG je lahko v zgodnjih fazah bolezni zvišana, v poznejših fazah pa znižana. Morda je zvišanje koncentracije IgM in G neke vrste kompenzacijska reakcija kot odziv na pomanjkanje IgA.
Funkcionalna aktivnost sistema T-limfocitov je zmanjšana; opazimo selektivno zmanjšanje števila B-limfocitov, odgovornih za sintezo Ig A, fagocitna povezava imunosti je motena, predvsem zaradi motenj kemotaksije in znotrajceličnih prebavnih procesov v nevtrofilcih.
Pri pregledu biopsije ledvice pri bolnikih z Alportovim sindromom podatki elektronske mikroskopije razkrivajo ultrastrukturne spremembe v glomerularni bazalni membrani: tanjšanje, motena struktura in cepitev glomerularnih bazalnih membran s spremembo njihove debeline in neenakomernimi konturami. V zgodnjih fazah dednega nefritisa okvara določa tanjšanje in krhkost glomerularnih bazalnih membran.
Stanjšanje glomerularnih membran je ugodnejši znak in je pogostejši pri deklicah. Bolj stalen elektronsko-mikroskopski znak pri dednem nefritisu je cepitev bazalne membrane, resnost njenega uničenja pa je povezana z resnostjo procesa.
Simptomi Alportovega sindroma pri otrocih
Prvi simptomi Alportovega sindroma v obliki izoliranega urinskega sindroma se najpogosteje odkrijejo pri otrocih prvih treh let življenja. V večini primerov se bolezen odkrije naključno. Urinarni sindrom se odkrije med preventivnim pregledom otroka, pred sprejemom v otroško ustanovo ali med akutnimi respiratornimi virusnimi okužbami (ARVI). V primeru patologije v urinu med ARVI. Pri dednem nefritisu, za razliko od pridobljenega glomerulonefritisa, ni latentnega obdobja.
V začetni fazi bolezni otrokovo zdravje malo trpi, značilna lastnost pa je vztrajnost in odpornost urinskega sindroma. Eden glavnih znakov je hematurija različnih stopenj resnosti, ki jo opazimo v 100 % primerov. Povečanje stopnje hematurije opazimo med ali po okužbah dihal, telesni aktivnosti ali po preventivnih cepljenjih. Proteinurija v večini primerov ne presega 1 g / dan, na začetku bolezni je lahko nestalna, z napredovanjem procesa pa se proteinurija povečuje. Občasno je lahko v urinu prisotna levkociturija s prevlado limfocitov, kar je povezano z razvojem intersticijskih sprememb.
Nato se delno poslabša delovanje ledvic, bolnikovo splošno stanje se poslabša: pojavijo se zastrupitev, mišična oslabelost, arterijska hipotenzija, pogosto okvara sluha (zlasti pri dečkih) in včasih okvara vida. Zastrupitev se kaže z bledico, utrujenostjo in glavoboli. V začetni fazi bolezni se izguba sluha v večini primerov odkrije le z avdiografijo. Izguba sluha pri Alportovem sindromu se lahko pojavi v različnih obdobjih otroštva, najpogosteje pa se izguba sluha diagnosticira v starosti 6-10 let. Izguba sluha pri otrocih se začne z visokimi frekvencami, doseže znatno stopnjo v zračni in kostni prevodnosti, prehaja pa iz izgube sluha za prevajanje zvoka v izgubo sluha za zaznavanje zvoka. Izguba sluha je lahko eden prvih simptomov bolezni in lahko predhodi urinarnemu sindromu.
V 20 % primerov imajo bolniki z Alportovim sindromom spremembe v vidnih organih. Najpogosteje odkrite anomalije so anomalije leče: sferofokija, sprednji, zadnji ali mešani lentikonus in različne vrste katarakte. V družinah z Alportovim sindromom je pomembna pogostost kratkovidnosti. Številni raziskovalci v teh družinah nenehno opažajo bilateralne perimakularne spremembe v obliki svetlih belkastih ali rumenkastih granulacij v rumenem telesu. Menijo, da je ta znak stalen simptom, ki ima visoko diagnostično vrednost pri Alportovem sindromu. KS Chugh in sodelavci (1993) so v oftalmološki študiji pri bolnikih z Alportovim sindromom ugotovili zmanjšanje ostrine vida v 66,7 % primerov, sprednji lentikonus v 37,8 %, pege na mrežnici v 22,2 %, katarakto v 20 % in keratokonus v 6,7 %.
Pri nekaterih otrocih z dednim nefritisom, zlasti pri razvoju ledvične odpovedi, opazimo znatno zaostajanje v telesnem razvoju. Z napredovanjem ledvične odpovedi se razvije arterijska hipertenzija. Pri otrocih jo pogosteje odkrijemo v adolescenci in starejših starostnih skupinah.
Za bolnike z dednim nefritisom je značilna prisotnost različnih (več kot 5-7) stigm vezivnotkivne dismorfogeneze. Med stigmami vezivnotkiva pri bolnikih so najpogostejše hipertelorizem oči, visoko nebo, anomalije ugriza, nenormalna oblika ušes, ukrivljenost mezinca na rokah in "sandalska vrzel" na stopalih. Za dedni nefritis je značilna enakomernost stigm dismorfogeneze znotraj družine, pa tudi visoka pogostost njihove razširjenosti med sorodniki probandov, po katerih liniji se bolezen prenaša.
V zgodnjih fazah bolezni se zazna izolirano zmanjšanje delnih ledvičnih funkcij: transport aminokislin, elektrolitov, koncentracijska funkcija, acidogeneza, kasnejše spremembe vplivajo na funkcionalno stanje tako proksimalnih kot distalnih delov nefrona in so značilne za kombinirane delne motnje. Zmanjšanje glomerularne filtracije se pojavi kasneje, pogosteje v adolescenci. Z napredovanjem dednega nefritisa se razvije anemija.
Za dedni nefritis je torej značilen stopenjski potek bolezni: najprej latentna faza ali skriti klinični simptomi, ki se kažejo z minimalnimi spremembami urinarnega sindroma, nato pa pride do postopne dekompenzacije procesa z zmanjšanjem delovanja ledvic z izraženimi kliničnimi simptomi (zastrupitev, astenija, razvojni zaostanek, anemija). Klinični simptomi se običajno pojavijo ne glede na plast vnetne reakcije.
Dedni nefritis se lahko manifestira v različnih starostnih obdobjih, kar je odvisno od delovanja gena, ki je do določenega časa v potlačenem stanju.
Klasifikacija
Obstajajo tri vrste dednega nefritisa
- Možnost I - klinično se kaže kot nefritis s hematurijo, izgubo sluha in okvaro oči. Potek nefritisa je progresiven z razvojem kronične ledvične odpovedi. Vrsta dedovanja je dominantna, povezana s kromosomom X. Morfološko se razkrije kršitev strukture bazalne membrane, njeno tanjšanje in cepljenje.
- Možnost II - klinično se kaže kot nefritis s hematurijo brez izgube sluha. Potek nefritisa je progresiven z razvojem kronične ledvične odpovedi. Vrsta dedovanja je dominantna, vezana na kromosom X. Morfološko se zazna tanjšanje bazalne membrane glomerularnih kapilar (zlasti laminadense).
- Možnost III - benigna družinska hematurija. Potek je ugoden, kronična ledvična odpoved se ne razvije. Vrsta dedovanja je avtosomno dominantna ali avtosomno recesivna. Pri avtosomno recesivni vrsti dedovanja je pri ženskah opazen hujši potek bolezni.
Diagnoza Alportovega sindroma
Predlagana so naslednja merila:
- prisotnost vsaj dveh bolnikov z nefropatijo v vsaki družini;
- hematurija kot vodilni simptom nefropatije pri probandu;
- prisotnost izgube sluha pri vsaj enem družinskem članu;
- razvoj kronične ledvične odpovedi pri enem ali več sorodnikih.
Pri diagnostiki različnih dednih in prirojenih bolezni se veliko mesto posveča celovitemu pristopu k pregledu in predvsem pozornosti podatkom, pridobljenim pri sestavljanju otrokovega rodovnika. Diagnoza Alportovega sindroma velja za veljavno v primerih, ko so pri bolniku odkriti 3 od 4 tipičnih znakov: prisotnost hematurije in kronične ledvične odpovedi v družini, prisotnost nevrosenzorične izgube sluha, patologija vida pri bolniku, odkrivanje znakov cepitve glomerularne bazalne membrane s spremembo njene debeline in neenakomernimi konturami med elektronsko mikroskopskimi značilnostmi biopsije.
Pregled bolnika mora vključevati klinične in genetske raziskovalne metode; ciljno preučevanje anamneze bolezni; splošni pregled bolnika ob upoštevanju diagnostično pomembnih meril. V fazi kompenzacije je mogoče patologijo odkriti le s poudarkom na sindromih, kot so prisotnost dedne obremenitve, hipotenzija, več stigm disembriogeneze, spremembe v urinarnem sindromu. V fazi dekompenzacije se lahko pojavijo zunajledvični simptomi, kot so huda zastrupitev, astenija, zaostanek v telesnem razvoju, anemija, ki se kažejo in stopnjujejo s postopnim zmanjševanjem delovanja ledvic. Pri večini bolnikov z zmanjšanim delovanjem ledvic opazimo naslednje: zmanjšano acido- in aminogenezo; 50 % bolnikov opazi znatno zmanjšanje sekretorne funkcije ledvic; omejen obseg nihanj optične gostote urina; motnje filtracijskega ritma in nato zmanjšanje glomerularne filtracije. Stadij kronične ledvične odpovedi se diagnosticira, ko imajo bolniki povišano raven sečnine v krvnem serumu (več kot 0,35 g/l) 3-6 mesecev ali več in zmanjšanje glomerularne filtracije na 25 % norme.
Diferencialno diagnozo dednega nefritisa je treba izvajati predvsem s hematurično obliko pridobljenega glomerulonefritisa. Pridobljeni glomerulonefritis ima najpogosteje akutni začetek, obdobje 2-3 tednov po okužbi, ekstrarenalne znake, vključno s hipertenzijo od prvih dni (pri dednem nefritisu, nasprotno, hipotenzija), zmanjšano glomerularno filtracijo na začetku bolezni, brez okvare delnih tubularnih funkcij, medtem ko so pri dednem prisotne. Pridobljeni glomerulonefritis se pojavi z izrazitejšo hematurijo in proteinurijo, s povečano sedimentacijo eritromiksa (ESR). Tipične spremembe v glomerularni bazalni membrani, značilne za dedni nefritis, so diagnostično pomembne.
Diferencialna diagnoza od dismetabolične nefropatije se izvaja s kronično ledvično odpovedjo, v družini pa so klinično odkrite heterogene ledvične bolezni, pri čemer je lahko prisoten spekter nefropatije od pielonefritisa do urolitiaze. Otroci se pogosto pritožujejo nad bolečinami v trebuhu in občasno med uriniranjem, v usedlini urina - oksalati.
Če obstaja sum na dedni nefritis, je treba bolnika napotiti na specializiran nefrološki oddelek, da se pojasni diagnoza.
Kaj je treba preveriti?
Katere teste so potrebne?
Koga se lahko obrnete?
Zdravljenje Alportovega sindroma
Režim vključuje omejitve težke telesne napore in bivanja na svežem zraku. Prehrana je polnovredna, z zadostno količino beljakovin, maščob in ogljikovih hidratov, ob upoštevanju delovanja ledvic. Zelo pomembno je odkrivanje in zdravljenje kroničnih žarišč okužbe. Uporabljajo se naslednja zdravila: ATP, kokarboksilaza, piridoksin (do 50 mg/dan), karnitin klorid. Tečaji se izvajajo 2-3-krat letno. Pri hematuriji se predpisujejo zeliščna zdravila - kopriva, sok aronije, rman.
V tuji in domači literaturi obstajajo poročila o zdravljenju s prednizolonom in uporabi citostatikov. Vendar je učinek težko oceniti.
Pri kronični odpovedi ledvic se uporabljata hemodializa in presaditev ledvic.
Za dedni nefritis ni metod specifične (učinkovite patogenetske) terapije. Vsi terapevtski ukrepi so namenjeni preprečevanju in upočasnjevanju upada delovanja ledvic.
Prehrana mora biti uravnotežena in visokokalorična, pri čemer je treba upoštevati funkcionalno stanje ledvic. Če ni funkcionalnih motenj, mora otrokova prehrana vsebovati dovolj beljakovin, maščob in ogljikovih hidratov. Ob prisotnosti znakov ledvične disfunkcije je treba količino beljakovin, ogljikovih hidratov, kalcija in fosforja omejiti, kar upočasni razvoj kronične ledvične odpovedi.
Telesno aktivnost je treba omejiti; otrokom je priporočljivo, da se izogibajo športu.
Izogibati se je treba stiku z nalezljivimi bolniki, zmanjšati tveganje za razvoj akutnih bolezni dihal. Sanacija žarišč kronične okužbe je potrebna. Preventivna cepljenja se pri otrocih z dednim nefritisom ne izvajajo, cepljenje je možno le zaradi epidemioloških indikacij.
Hormonsko in imunosupresivno zdravljenje pri dednem nefritisu ni učinkovito. Obstajajo znaki pozitivnega učinka (zmanjšanje proteinurije in upočasnitev napredovanja bolezni) pri dolgotrajni večletni uporabi ciklosporina A in zaviralcev ACE.
Pri zdravljenju bolnikov se uporabljajo zdravila, ki izboljšujejo presnovo:
- piridoksin - 2-3 mg/kg/dan v 3 odmerkih 4 tedne;
- kokarboksilaza - 50 mg intramuskularno vsak drugi dan, skupaj 10-15 injekcij;
- ATP - 1 ml intramuskularno vsak drugi dan, 10-15 injekcij;
- vitamin A - 1000 ie/leto/dan v 1 odmerku 2 tedna;
- Vitamin E - 1 mg/kg/dan v 1 odmerku 2 tedna.
Ta vrsta terapije pomaga izboljšati splošno stanje bolnikov, zmanjšati tubularne disfunkcije in se izvaja v tečajih 3-krat letno.
Levamizol se lahko uporablja kot imunomodulator - 2 mg/kg/dan 2-3-krat na teden s premori med odmerki 3-4 dni.
Glede na raziskovalne podatke ima hiperbarična oksigenacija pozitiven učinek na resnost hematurije in ledvične disfunkcije.
Najučinkovitejša metoda zdravljenja dednega nefritisa je pravočasna presaditev ledvice. V tem primeru ne pride do ponovitve bolezni v presajeni ledvici; v majhnem odstotku primerov (približno 5 %) se lahko v presajeni ledvici razvije nefritis, povezan z antigeni glomerularne bazalne membrane.
Obetavna smer je prenatalna diagnostika in gensko inženirska terapija. Poskusi na živalih kažejo visoko učinkovitost prenosa normalnih genov, odgovornih za sintezo alfa verig kolagena tipa IV, v ledvično tkivo, po katerem opazimo sintezo normalnih kolagenskih struktur.
Napoved
Prognoza za dedni nefritis je vedno resna.
Prognostično neugodna merila za potek dednega nefritisa so:
- moški spol;
- zgodnji razvoj kronične ledvične odpovedi pri družinskih članih;
- proteinurija (več kot 1 g/dan);
- odebelitev glomerularnih bazalnih membran po mikroskopiji;
- akustični nevritis;
- delecija v genu Col4A5.
Prognoza za benigno familiarno hematurijo je ugodnejša.